
We use a number of evolutionary techniques where molecular structures can “evolve” through virtual mutation and selection based on fitness criteria (e.g., drug-likeness, binding affinity). It is a way of creating new virtual molecules but with certain restrictions, such as limiting the molecular weight and molecular complexity ranges, and “growing them” into new virtual chemical structures. Various approaches are used for creating new virtual molecules and “growing them” into new virtual chemical structures:
Evolutionary Molecular Design (EMD)
- A machine-learning-assisted method for generating new chemical entities by iterating structural modifications.
De Novo Molecular Design
- An approach using evolutionary strategies to generate entirely new molecular structures optimized for specific biological or chemical properties.
Computational Mutagenesis
- A strategy where molecular fragments undergo systematic changes (mutation, recombination) to explore novel chemical spaces.
Below is a dynamic example of molecular evolution:
Starting with an alkaloid (compound A) on top as the “seed compound” we create a selection of virtual molecules based on various evolutionary fitness criteria that progresses (middle image) to a difluorinated derivative (compound B) on bottom.
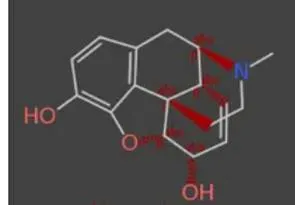
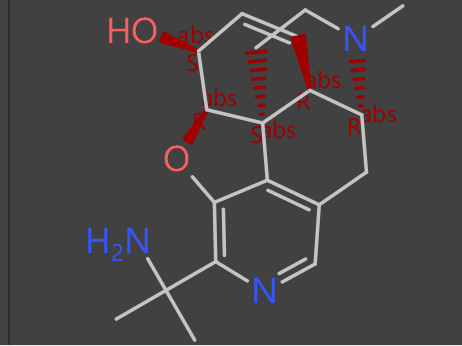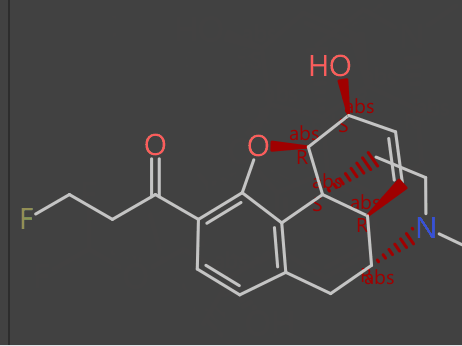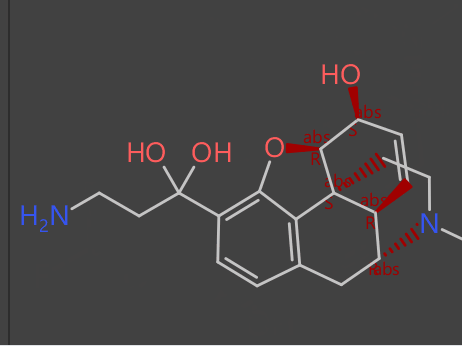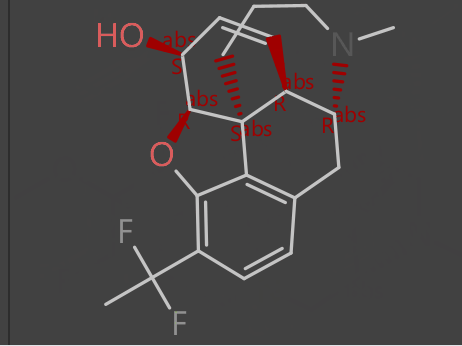
The evolutionary phase can generate 1000s or billions of potential new virtual compounds which can be probed further to find useful and interesting functionality.
How can this be used?
Some of our Services include:
Virtual Library Creation
Processing and generating large collections of computationally designed molecules for use in, for example, drug discovery or the discovery of new nanomaterials. This is achieved using cheminformatics tools, combinatorial chemistry, and often AI-driven methods.
As part of this work, we offer the formation of boutique curated datasets and also data cleaning of existing virtual libraries. Key steps include:
- Scaffold Design – Selecting core structures based on known bioactive molecules.
- Molecular Enumeration – Generating diverse analogs by modifying functional groups.
- Filtering & Prioritization – Applying physicochemical and ADMET filters to select drug-like candidates.
- Database Storage & Screening – Organizing molecules for virtual screening against biological targets.
Pharmacophore Scaffold Hopping and Target Hopping
Pharmacophore scaffold hopping involves modifying a molecule’s core structure (scaffold) while retaining key functional groups essential for biological activity. This strategy helps discover novel compounds with improved properties, such as enhanced potency, selectivity, or pharmacokinetics.
Target hopping refers to designing compounds that interact with different biological targets while maintaining a similar pharmacophore. This approach aids in discovering new therapeutic applications (drug repurposing) and reducing drug resistance.
Benefits of both strategies include expanding chemical diversity, overcoming patent barriers, improving drug-likeness, and identifying alternative treatments for diseases, making them essential in drug discovery and development.
Activity Cliffs Analysis
This is a powerful technique primarily used for finding outliers from a dataset. Examples include applying the technique to economic issues such as finding huge price differences for very similar compounds for chemical vendors or food ingredient companies, or identifying unusually high actives from a dataset of similar compounds.
We also undertake related work such as 3D activity cliffs and pharmacophore activity cliffs. By identifying activity cliffs, we can then probe further to understand why they are occurring and use this knowledge to develop new insights and make new discoveries.
Pattern Recognition Studies
Pattern recognition studies in drug discovery involve identifying trends and relationships within chemical and biological datasets to uncover new drug leads. They can also be used to analyze chemical ecological interactions and aid in the study of “chemical systematics.”
Techniques like t-distributed Stochastic Neighbor Embedding (t-SNE) and Self-Organizing Maps (SOMs) help visualize complex, high-dimensional data by grouping similar compounds based on structural or activity-based features. Other clustering algorithms (e.g., k-means, hierarchical clustering) classify molecules into meaningful groups, aiding scaffold hopping and lead optimization.
By analyzing both existing and novel datasets, these methods reveal hidden patterns, predict bioactivity, and accelerate hit identification, ultimately guiding rational drug design and reducing experimental costs.
Similarity Searches
Similarity searches involve screening public, internal, or proprietary databases to identify compounds structurally or functionally similar to a reference molecule.
This process integrates various filtering techniques, such as restricting results within a specific logP range (for lipophilicity control), exploring beyond the Rule of 5 (Ro5) for non-traditional drug-like compounds, and applying multi-component descriptors (e.g., molecular weight, hydrogen bond donors/acceptors, and topological indices) for precise selection.
These searches help identify novel scaffolds, optimize lead compounds, and support scaffold and target hopping strategies, accelerating the drug discovery process while ensuring desirable physicochemical and pharmacokinetic properties.
Small and Retired Datasets
Discovering new and valuable leads that others may overlook, enhancing drug and molecular discovery through precision-driven analysis rather than sheer data volume.